What Are ATP1A3-Related Disorders?
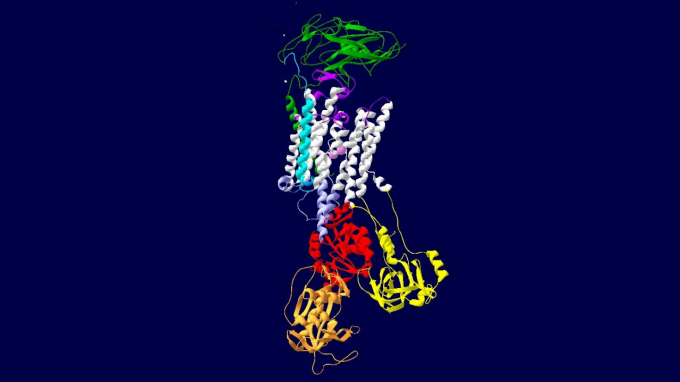
Variants in the ATP1A3 gene are associated with various neurological manifestations. Research team member Kathleen J. Sweadner, PhD, of Harvard University, imaged this ATP1A3 gene.
What do we mean when we say we study ATP1A3-related disorders? Well, we’re taking a deep dive into how disease-causing variants of the ATP1A3 gene cause a variety of movement disorders.
With ATP1A3-related neurologic disorders, at least three phenotypes have been delineated: rapid-onset dystonia-parkinsonism (RDP); alternating hemiplegia of childhood (ACH); and cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS).
(That being said, some affected individuals have intermediate phenotypes or only a few features that do not fit well into one of these major phenotypes.)
Let’s discuss what all of this means.
Rapid-Onset Dystonia-Parkinsonism (RDP)
RDP is a movement disorder, characterized by the abrupt onset of parkinsonism and dystonia (which makes muscles contract involuntarily and can cause repetitive or twisting movements).
Often fever, physiologic stress, or alcohol binges trigger the onset of symptoms. After their initial appearance, symptoms often stabilize with little improvement; occasionally second episodes occur with abrupt worsening of symptoms.
Anxiety, depression and seizures have been reported. Age of onset ranges from four to 55 years, although a childhood variation of RDP with onset between ages nine and 14 months has been reported.
Alternating Hemiplegia of Childhood (ACH)
AHC is a complex neurodevelopmental syndrome most frequently manifesting in infancy or early childhood.
Classic AHC causes recurrent episodes of paralysis that involve one or both sides of the body, multiple limbs, or a single limb.
The paralysis may affect different parts of the body at different times and may be brief or last for several days. A characteristic feature of AHC is that symptoms disappear during sleep and return upon waking. Many affected children display some degree of developmental delay, abnormal eye movements, uncontrolled limb movements and seizures.
Triggers including psychological stress / excitement; environmental stressors (e.g., bright light, excessive heat or cold, excessive sound, crowds); water exposure (e.g., bathing, swimming); certain foods or odors (e.g., chocolate, food dyes); missed meals; excessive or atypically strenuous exercise; illness; irregular sleep (missing a nap, delayed bedtime).
CAPOS (Cerebellar Ataxia, Areflexia, Pes Cavus, Optic Atrophy, and Sensorineural Hearing Loss)
CAPOS (cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss) syndrome is characterized by episodes of ataxic encephalopathy and/or weakness during and after a febrile illness.
Onset is between ages six months and four years. Some acute symptoms resolve; progression of sensory losses and severity vary.
Most people with CAPOS syndrome have one to three episodes during their lifetime. Signs and symptoms during an episode may include low muscle tone, unusual eye movements (nystagmus or strabismus), problems with speech (dysarthria), difficulty swallowing (dysphagia), reduced or absent reflexes, and hearing loss.
Some people may lose consciousness or go into a coma during an episode. Though many of the signs and symptoms of CAPOS syndrome get better as the fever and illness improve, some symptoms, including movement problems, may continue.